Tutorial of RNA-seq tumor immunity analysis
2021-08-19
Chapter 1 Introduction
This is a tutorial about integrative computing analysis of tumor immunity using bulk RNA-sequencing (RNA-seq) data. We will focus on inferring immune infiltration, immune repertoire, immune response and neoantigen prediction from a gene expression profile.
We developed a RNA-seq immune analysis pipeline named RIMA that is available at https://github.com/liulab-dfci/RIMA/.
Tumor RNA-seq has become an important technique for molecular profiling and
immune characterization of tumors. RNA-seq Immune Analysis (RIMA) performs
integrative computational modeling of the tumor microenvironment from bulk tumor
RNA-seq data, which has the potential to offer essential insights to cancer
immunology and immune-oncology studies.
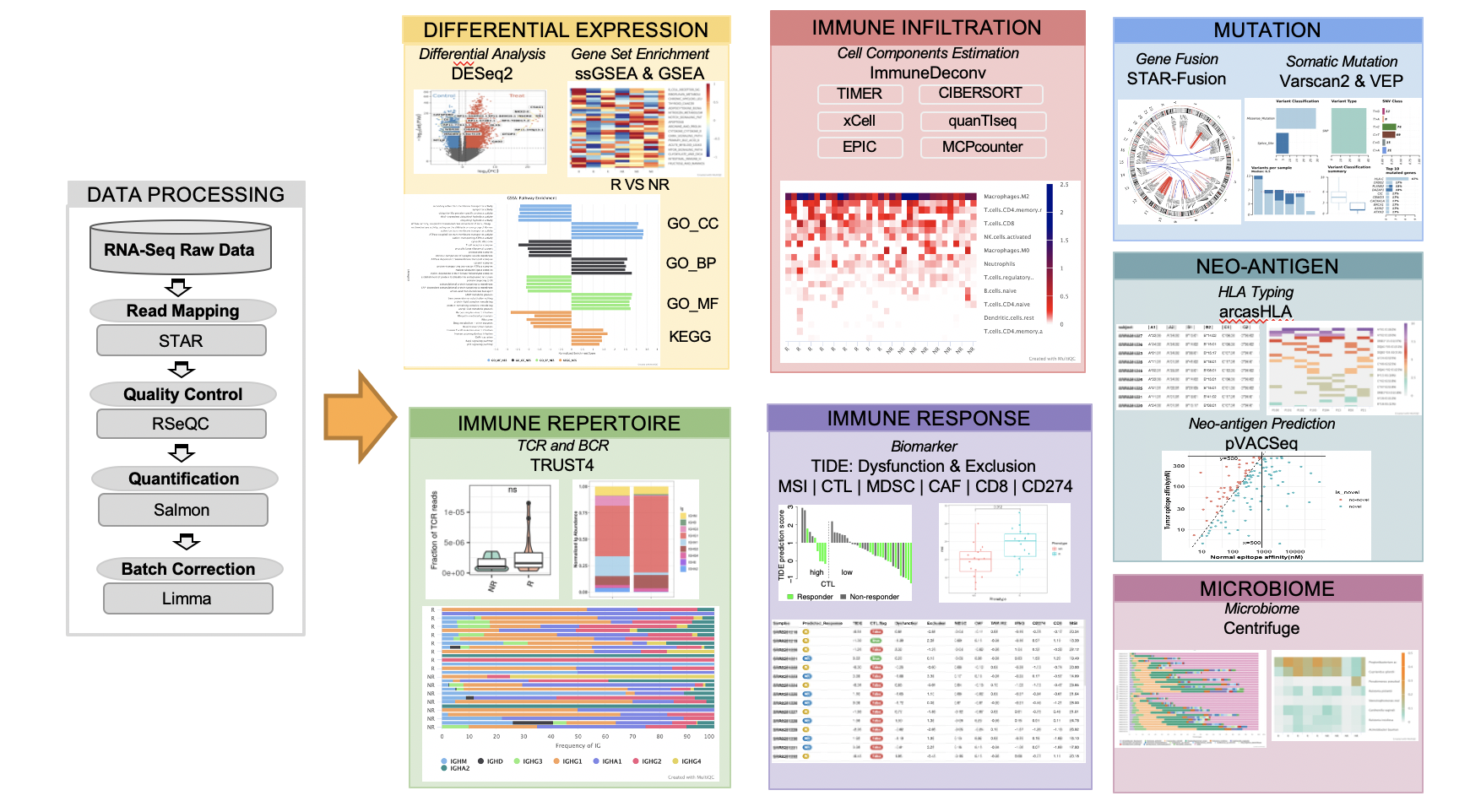
Figure 1.1: Flowchat of RIMA pipeline
- Read mapping
- Quality control
- Gene quantification
- Batch effect removal
- Differential expression analysis
- Immune repertoire inference
- Immune infiltration estimation
- Immunotherapy response prediction
- Gene fusion
- Microbiome characterization
- Neoantigen detection
Available Tools Checklist
| Methods | Description | |
|---|---|---|
| —PREPROCESSING— | ||
| STAR | Spliced Transcript Alignment to a Reference | |
| Salmon | Gene Quantification | |
| RSeQC | High Throughput Sequence Data Evaluation | |
| batch_removal | Remove Batch Effects Using Limma | |
| —DIFFERENTIAL EXPRESSION— | ||
| DESeq2 | Gene Differential Expression Analysis | |
| GSEA | Gene Set Enrichment Analysis | |
| ssGSEA | Single-sample GSEA | |
| —IMMUNE REPERTOIRE— | ||
| TRUST4 | TCR and BCR Sequence Analysis | |
| —IMMUNE INFILTRATION— | ||
| ImmuneDeconv | Cell Components Estimation | |
| —IMMNUE RESPONSE— | ||
| MSIsensor2 | Microsatellite Instability (MSI) Detection | |
| TIDEpy | T cell dysfunction and exclusion prediction | |
| —FUSION— | ||
| STAR-Fusion | Identify the fusion gene pairs | |
| —MICROBIOME— | ||
| Centrifuge | Bacterial Abundance Detection | |
| —NEO-ANTIGEN— | ||
| arcasHLA | HLA Class I and Class II Genotyping |